Ein Molekül läßt sich als tiefgeschachtelte diskrete Struktur auffassen, wobei dabei verschiedene Eigenschaften und Beschreibungen in Betracht gezogen werden können - Topologie, räumliche Anordnung etc. Mit unserem Verfahren läßt sich leicht die Symmetrie eines Moleküls unter Berücksichtigung der jeweiligen Beschreibung bestimmen.
Damit sind topologische und dreidimensionale Voll- und Teilstruktursuchen möglich. Die Algorithmen sind besonders effektiv, da ihre Komplexität nicht fest ist, sondern sich individuell dem Aufwand anpaßt, ein einzelnes Molekül zu identifizieren.
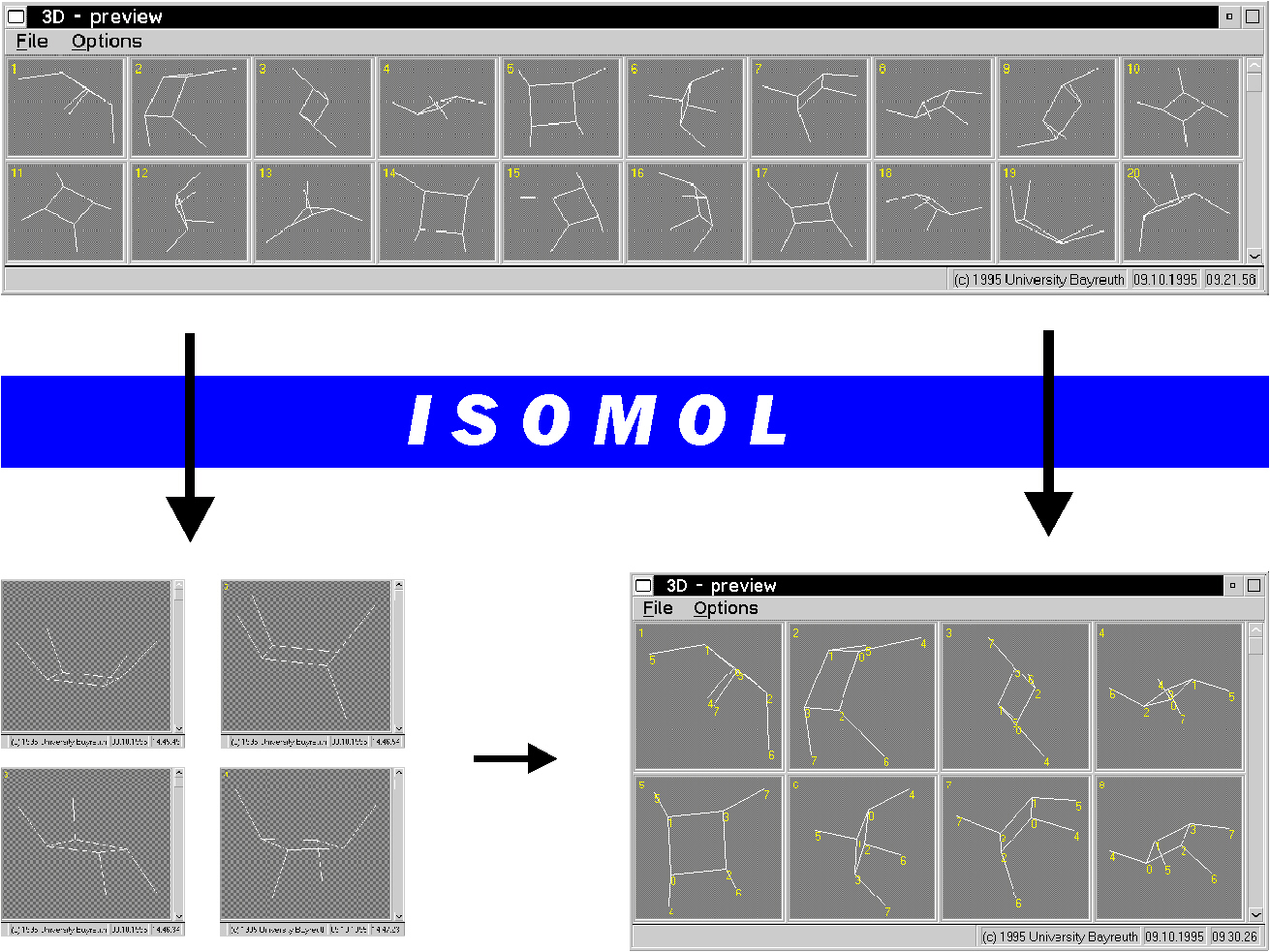
Darüber hinaus ergibt sich damit auch ein neues Verfahren zur kanonischen Numerierung räumlicher Molekülmodelle (vorgestellt auf der 2. Elektronischen Konferenz zur Computerchemie [3]).
Die Aufgabe, eine Menge räumlicher Anordnungen eines Moleküls in Ähnlichkeitsklassen einzuteilen, kann also vollautomatisch gelöst werden (s. die Abbildung mit Tetramethylcyclobutan).
{kind=link}